甲烷,作为天然气的主要成分,不仅是一种清洁的能源,也是合成多种高附加值化学品的重要原料。然而,由于其分子结构的稳定性,甲烷的化学转化过程通常需要高温高压条件,这不仅消耗大量能源,还可能引发环境问题。为了解决这一挑战,光催化技术应运而生,它利用太阳能在温和条件下激活甲烷,为化学合成提供了一种可持续的凯发官网首页的解决方案。尽管这一领域充满潜力,但目前仍面临诸多技术难题。
在传统的甲烷转化过程中,如蒸汽甲烷重整,虽然实现了甲烷的大规模工业应用,但这一过程不仅能耗高,还伴随着大量的二氧化碳排放。因此,探索在温和条件下直接转化甲烷的方法,对于提高能源利用效率和减少环境污染具有重要意义。[1-3]
近年来,光催化技术因其在温和条件下活化甲烷的能力而受到广泛关注。光催化甲烷转化利用光能激发催化剂,产生高能电子-空穴对,这些高能电子-空穴对能够打破甲烷分子的化学惰性,从而在较低的温度和压力下实现甲烷的高效转化。[4-6]这一过程不仅能够降低能耗,还能减少温室气体排放,对于推动绿色化学合成具有重要的战略意义。
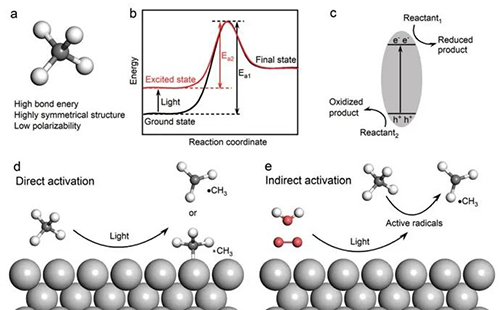
甲烷以其高度的化学稳定性而著称,这种稳定性源自其对称的四面体几何结构,其中四个等价的c-h键赋予了它高键能、低电子及质子亲和力和低极化性(见图1a)。[7,8]此外,甲烷的最高占据分子轨道(homo)与最低未占据分子轨道(lumo)之间的能隙较大,这增加了它在分子层面上获得或失去电子的难度。[1,9]这种内在的化学惰性意味着在热催化转化过程中,需要逾越较高的活化能障碍(ea),通常需要在700至1000摄氏度的高温下进行,以实现有效的转化。尽管多年来科学家们不懈努力开发新型催化剂以提高甲烷转化的效率,但热催化过程的高能耗和碳排放问题依然存在。目前,唯一大规模应用的甲烷转化技术是将其通过蒸汽重整间接转化为合成气,进而生产烯烃、甲醇和液态烃。而将甲烷高效转化为一系列含碳化学品,包括醇类、芳香烃、长链烷烃和烯烃,一直是催化化学领域追求的目标。[10]
非均相光催化剂继电子-空穴分离和转移过程之后,主要涉及催化剂-介质界面上发生的过程,包括反应物的吸附与活化、中间体的生成以及产物的解吸与脱附等。[11-13]催化界面上的复杂反应过程对精细调节催化反应的总效率提出了挑战。[14,15]甲烷活化通常被认为是甲烷转化反应中的速率控制步骤。[16,17]基于非均相催化剂的光催化甲烷活化可分为两类:
1)直接活化,即甲烷在光照条件下直接吸附在光催化剂表面被活化(图 1d);
2)间接活化,即甲烷在水和氧分子等其他反应物的光诱导活性自由基的帮助下被活化(图 1e)。
尽管两种甲烷活化途径之间存在差异,但光诱导产生的活性氧,如产生光生表面活性位点(氧化物上的o⁻)和活性自由基(•oh和•o²⁻)在以上叙述的两种途径中都发挥着关键作用。
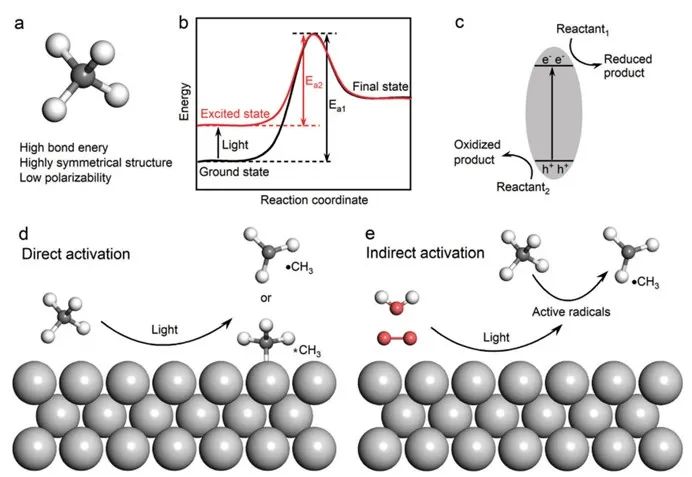
图1 a)甲烷的分子结构。b)基态和激发态反应中的能量变化。c)光催化反应的基本原理。d)甲烷在光催化剂表面的直接活化。e)光诱导活性自由基对甲烷的间接活化。
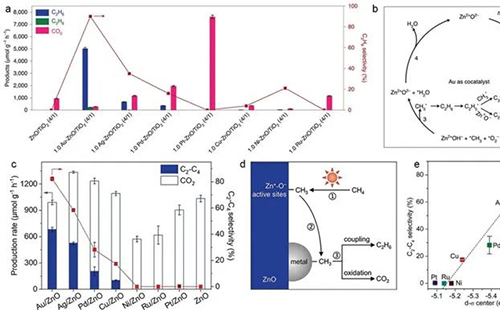
对于直接活化途径而言,为优化光催化甲烷活化,通常改性策略是酸/碱位点和晶体缺陷工程改性策略。[18-20]金属氧化物上的碱性位点被认为在甲烷活化中起着至关重要的作用,因为它们与弱酸性甲烷具有相对较强的电子相互作用。[21-23]吸附在金属氧化物上的甲烷会形成与金属阳离子配位的负电基团和与金属氧化物的碱性晶格氧配位的正电基团,二者作用使金属氧化物表面的 c–h 键极化(图 2a)。根据金属氧化物的碱性强弱,这两部分将显示路易斯酸(正)或路易斯碱(负)。[24]此外,纳米催化剂中的缺陷导致晶胞平移对称性的破坏,包括三维体积缺陷(如孔隙)、二维平面缺陷(如晶界)、一维线缺陷(位错)和零维点缺陷(如空位)。这些缺陷直接影响金属氧化物的配位结构和相应的催化性能。[25]例如,pd修饰的zno-au复合材料具有独特的界面缺陷结构,促进ch₄分子解离为甲氧基和甲基,促进光催化甲烷-乙烯转化(图2b)。[26]
对于间接途径来说,氢原子转移是激活甲烷 c–h 键的有效策略。它是涉及通过自由基反应中质子和电子的分离而转移氢原子(图 2c)的一个基本反应。[27,28]光催化氢原子转移是利用光催化过程中产生的自由基中间体来活化 c–h 键。[29]气相中的裸露的 (cuo)⁺ 阳离子可以与以氧为中心的自由基相互作用,促进氢原子转移和甲烷活化。[30]据报道,基于同步辐射的原位光催化质谱技术可捕获光催化甲烷氧化反应中的气相活性中间体。[31]除了能够检测到 co₂、h₂o、c₂h₆ 和 ch₃oh 等几种稳定物质外,还能够检测到活性甲基自由基(•ch₃)和甲氧基自由基(ch₃o•)(图 2d)。此外,质谱结合量子化学计算显示,(auo)⁺ 通过选择性地从 (auo)⁺ 中转移氧原子,而不是从甲烷中提取氢原子转移氢原子,将甲烷活化为甲醇,尽管在同系物 (cuo)⁺ 和 (ago)⁺ 中观察到了后一种情况。
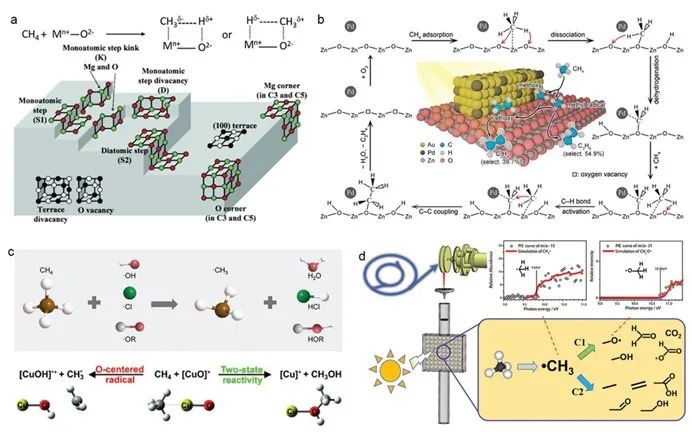
图 2. a) 甲烷在碱性金属氧化物表面的吸附和 c–h 键极化以及 mgo 表面不同位置的示意图。[21,22] b) 在 zno-aupd 混合催化剂的情况下,通过表面烷氧基中间体将 ch₄ 光催化转化为 c₂h₄ 的示意图。[26] c) 通过氢原子转移活化甲烷的示意图。[30] d) 通过原位同步辐射光电离质谱法检测光催化甲烷氧化反应过程中的反应中间体。[31]
甲烷的氧化脱氢反应是另一种广泛研究的温和条件下甲烷活化和转化的间接途径[32]。根据涉及到的氧活性物质的形式,甲烷的氧化脱氢反应主要遵循三种机理:rideal-eley (r-e)机理和langmuir-hinshelwood (l-h)机理,二者均以表面吸附氧为主,以及mars-van krevelen (mv-k)机理,是以晶格氧为主。下面简单介绍一下这些反应机理:
1)r-e机理:ch₄分子最初与过渡金属氧化物中的晶格氧结合,导致ch₄分子中的c–h键断裂并形成自由甲基自由基,随后甲基自由基被氧化。[33]同时,结合过程会消耗晶格中的氧,从而形成弱电氧空位。这些空位吸收分子氧来补充失去的晶格氧,从而完成催化循环(图3a)。
2)l-h机理:催化剂表面对分子氧的化学吸附明显比对甲烷的化学吸附容易,这导致分子氧在暴露于含氧大气中时优先吸附在催化剂表面[34]与分子氧相比,这些被吸附的氧物种与ch₄分子结合时表现出更高的反应活性,导致甲烷中c–h键被拉长。这种拉长随后会破坏了ch₄分子的正四面体结构,产生活性甲基自由基,促进 ch₄的氧化脱氢(图3b)。l–h 机理主要用于贵金属负载的金属氧化物催化剂。[35]
3)mv-k机理涉及表面氧反应和晶格氧迁移。[36]首先,气态ch₄分子被吸附在催化剂的活性位点上。然后,吸附的ch₄分子与表面晶格氧发生反应,生成 ch₃⁺ 离子,随后被氧化成 co₂ 和 h₂o 分子等吸附产物,从而产生氧空位。这一步骤可称为催化剂还原。最后,吸附产物被解吸,而内部晶格氧迁移到表面,用表面吸附的氧重新填满氧空位,这就是催化剂的再氧化(图 3c)。
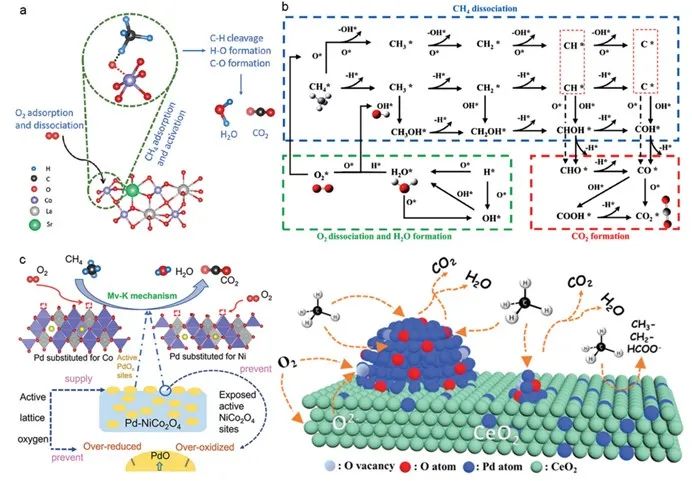
图3. a) 针对镧、钴基钙钛矿氧化物催化剂提出的超面 rideal-eley 机理示意图。[33]b) 通过 langmuir-hinshelwood 机理进行的甲烷催化燃烧反应途径。[35] c) 通过 mars-van krevelen 机理进行的甲烷氧化反应,其中 pd 掺入 pd-nico₂o₄ 和 pd/ceo₂。[36,37]


参考文献
[1]. n. j. gunsalus, a. koppaka, s. h. park, s. m. bischof, b. g. hashiguchi, r. a. periana, chem. rev. 2017, 117, 8521.
[2]. y. song, e. ozdemir, s. ramesh, a. adishev, s. subramanian, a. harale, m. albuali, b. a. fadhel, a. jamal, d. moon, s. h. choi, c. t. yavuz, science 2020, 367, 777.
[3]. y. chen, j. wei, m. s. duyar, v. v. ordomsky, a. y. khodakov, j. liu, chem. soc. rev. 2021, 50, 2337.
[4]. t. kong, y. jiang, y. xiong, chem. soc. rev. 2020, 49, 6579.
[5]. p. bellotti, h.-m. huang, t. faber, f. glorius, chem. rev. 2023, 123, 4237.
[6]. l. zhang, l. liu, z. pan, r. zhang, z. gao, g. wang, k. huang, x. mu, f. bai, y. wang, w. zhang, z. cui, l. li, nat. energy 2022, 7, 1042
[7]. p. tang, q. zhu, z. wu, d. ma, energy environ. sci. 2014, 7, 2580.
[8]. b. wang, s. albarracín-suazo, y. pagán-torres, e. nikolla, catal. today 2017, 285, 147.
[9]. h. schwarz, angew. chem., int. ed. 2011, 50, 10096.
[10]. r. g. bergman, nature 2007, 446, 391.
[11]. j. c. védrine, top. catal. 2002, 21, 97.
[12]. i. fechete, y. wang, j. c. védrine, catal. today 2012, 189, 2.
[13]. r. schlögl, angew. chem., int. ed. 2015, 54, 3465.
[14]. c. m. friend, b. xu, acc. chem. res. 2017, 50, 517.
[15]. x. cui, w. li, p. ryabchuk, k. junge, m. beller, nat. catal. 2018, 1, 385.
[16]. y. chen, x. mu, x. luo, k. shi, g. yang, t. wu, energy technol. 2020, 8, 1900750.
[17]. s. h. morejudo, r. zanón, s. escolástico, i. yuste-tirados, h. malerød-fjeld, p. k. vestre, w. g. coors, a. martínez, t. norby, j. m. serra, c. kjølseth, science 2016, 353, 563.
[18]. m. campanati, g. fornasari, a. vaccari, catal. today 2003, 77, 299.
[19]. a. vojvodic, j. k. nørskov, natl. sci. rev. 2015, 2, 140.
[20]. a. nilsson, l. g. m. pettersson, b. hammer, t. bligaard, c. h. christensen, j. k. nørskov, catal. lett. 2005, 100, 111.
[21]. v. choudhary, j. catal. 1991, 130, 411.
[22]. h. u. hambali, a. a. jalil, a. a. abdulrasheed, t. j. siang, t. a. t. abdullah, a. ahmad, d.-v. n. vo, int. j. energy res. 2020, 44, 5696.
[23]. j. ashok, z. bian, z. wang, s. kawi, catal. sci. technol. 2018, 8, 1730.
[24]. c. chizallet, g. costentin, m. che, f. delbecq, p. sautet, j. phys. chem. b 2006, 110, 15878.
[25]. w. jiang, j. low, k. mao, d. duan, s. chen, w. liu, c.-w. pao, j. ma, s. sang, c. shu, x. zhan, z. qi, h. zhang, z. liu, x. wu, r. long, l. song, y. xiong, j. am. chem. soc. 2021, 143, 269.
[26]. y. jiang, y. fan, s. li, z. tang, ccs chem. 2022, 5, 30.
[27]. b. an, q.-h. zhang, b.-s. zheng, m. li, y.-y. xi, x. jin, s. xue, z.-t. li, m.-b. wu, w.-t. wu, angew. chem., int. ed. 2022, 61, e202204661.
[28]. j. wang, l. zhang, d. zeng, w. wang, r. li, t. jia, b. cui, h. chu, w. wang, appl. catal., b 2023, 337, 122983.
[29]. n. dietl, c. van der linde, m. schlangen, m. k. beyer, h. schwarz, angew. chem., int. ed. 2011, 50, 4966.
[30]. c. liu, b. qian, t. xiao, c. lv, j. luo, j. bao, y. pan, angew. chem., int. ed. 2023, 62, e202304352.
[31]. s. zhou, j. li, m. schlangen, h. schwarz, angew. chem., int. ed. 2016, 55, 10877.
[32]. t. wang, c. zhang, j. wang, h. li, y. duan, z. liu, j. y. lee, x. hu, s. xi, y. du, s. sun, x. liu, j.-m. lee, c. wang, z. j. xu, j. catal. 2020, 390, 1.
[33]. x. bu, j. ran, j. niu, z. ou, l. tang, x. huang, mol. catal. 2021, 515, 111891.
[34]. e. becker, p.-a. carlsson, l. kylhammar, m. a. newton, m. skoglundh, j. phys. chem. c 2011, 115, 944.
[35]. t. wang, l. qiu, h. li, c. zhang, y. sun, s. xi, j. ge, z. j. xu, c. wang, j. catal. 2021, 404, 400.
[36]. s. chen, s. li, r. you, z. guo, f. wang, g. li, w. yuan, b. zhu, y. gao, z. zhang, h. yang, y. wang, acs catal. 2021, 11, 5666.
[37]. d. hu, v. v. ordomsky, a. y. khodakov, appl. catal., b 2021, 286, 119913.